1. Overview
Gene expression and gene co-expression changes over the lifespan and varies by sex, tissue, and cell type. Deviations from normal gene expression changes likely contribute to neuropsychiatric disorders. Change of gene co-expression, which may reflect regulatory relationships among genes, is also implied in the etiology of multiple psychiatric disorders. However, normal gene variation and co-expression in the human brain has yet to be fully quantified and defined. Many previous studies that were specific to human brain were carried out without checking whether the genes tested were expressed in brain.
Therefore, we developed a novel database called Brain gene EXPression (BrainEXP) (www.brainexp.org), which provides information about individual genes and related co-expression in normal human brains with filters of brain regions, age stages, and sex. BrainEXP stores data of 4,567 samples from 2,863 healthy individuals with 44 brain regions gathered from existing public databases and our own data. It serves as a reference dataset that can be used in studying gene expression changes in neuropsychiatric brains. When you search one gene or list of genes in the BrainEXP, it can offer you comprehensive information about expression levels and variance of specific candidate genes in specific brain regions at different ages, brain regions, and sex. You can also get the co-expression modules results analyzed by weighted gene co-expression network analysis. BrainEXP also supplies the gene annotation to your input gene list.
In the home page, you can search the database in gene name or symbol. You can choose the “Advanced” search to get the advanced search page, and then limit the factors of interest, the sex, brain regions, and platforms.
2. Data Summary
CurrentlyBrainEXP contains 4,567 normal human brain samples of 2,863 individuals with 56 brain regions from our own data and existing public databases.
☆ Analysis workflow
The overall of the analysis workflow shows below.

Figure S1. An overview of the analysis workflow for data collection and basic analysis.
Data pre-processing:
Data were collected from vastly different platforms and they are hardly comparable. We normalized the data based on the platform information by the same workflow.
■ HG-U133A (Human Genome U133A 2.0 Array) / HG-U133P (Human Genome U133 Plus 2.0 Array)The data pre-processing workflow of platforms HG-U133A and HG-U133P are the same, so we describe these platforms together. After we received the normal human brain gene expression raw data (.CEL files), we filtered the probes with a detection P value above 0.06 in at least 80 % samples, and then filtered the samples with a detection P value above 0.06 in at least 80 % probes. We processed the CEL files using standard tools available within the affy package in R. The CEL files were processed with the expression command to convert the raw probe intensities to probeset expression values. We used the CLL, CLLbatch, simpleaffy packages to obtain the quality control results of samples and filtered the poor quality samples. After that, we used the impute package in R to impute the NA values in the dataset. Then we used gene-based expression values to replace the probe-based values. In the case of multi-probe-per-gene, we took the max of the probe-based values as the representative values of the gene. After we logit transformed the data and corrected the batch information using ComBat, we put the data from different data sets together by ComBat method. We corrected batch effects using the ComBat, which adjusts for known batches using an empirical Bayesian framework (W.E. Johnson, 2007).
■ HG1.0 (HuGene-1_0-st) /HG1.1 (HuGene-1_1-st)The data pre-processing workflow is the same for both platforms HG1.0 and HG1.1, so we describe these platforms together. The raw data is also in the .CEL format, so we took the normal human brain CEL files to the expression console to get the gene expression matrixes. We removed the probesets that crosshyb-type=3, which means the probesets perfectly or partially match more than one sequence. We filtered the samples by hierarchical cluster with average linkage –if a sample (or small group of samples) didn’t cluster with other samples, we defined the sample as an outlier and removed it. After that, we used the impute package in R to impute the NA values in the dataset. Then we used gene-based expression values to replace the probe-based values. In case of multi-probe-per-gene, we took the max of them as the representative values of the gene. We then corrected batch effects to remove the batch confounder. Finally, we put the data from different data sets together by ComBat method.
■ Illumina Human 49K Oligo array (HEEBO-7 set) / Illumina Human HT-12 V3.0 expression beadchipThe data we received from these two datasets are both non-normalized data with similar data structure, so we preprocessed the data similarly. We filtered samples by hclust function in R (similar to how we processed the HG1.0 /HG1.1 data). In the case of multi-probe-per-gene, we took the max of the probe-based values as the representative values of the gene. After that, we use the ComBat function to adjust the batch effect within dataset and among datasets.
■ RNA-seq (Ribo-Zero)Table S1. The basic tools and workflow of the RNA-seq (RZ) dataset analysis.
| Workflows | Purpose | Tools |
|---|---|---|
| Quality Control | Generate quality statistics | FastQC v0.11.2 |
| Read Preprocessing | 1. Trim low quality bases | Trimmomatic v0.22 |
| 2. Clip adapters/primers | ||
| 3. Filter low quality reads | ||
| 4. Filter ambiguous reads | ||
| Align Reads to Reference Genome | Align trimmed fastq reads to transcript reference | TopHat v2.0.13 |
| Collect Multiple Metrics | Insert Size, Quality Score, Cycle, and RNA seq feature | Picard v1.109 |
| Assemble Transcripts and Quantification | Estimate RNA expression levels by FPKM using TopHat alignment results | Cufflinks v2.2.1 |
| Read counts using TopHat alignment results | HTSeq |
RNA-seq was performed using the Illumina TruSeq library construction protocol. This is a non-strand specific polyA+ selected library. The sequencing produced 76-bp paired end reads.Alignment to the HG19 human genome was performed using TopHat v1.4.1 assisted by the GENCODE v19 transcriptome definition. In post-processing, unaligned reads are reintroduced into the bam. The final bam contains aligned and unaligned reads, and marked duplicates. It should be noted that TopHat produces multiple mappings for some reads, but in post-processing one read is flagged as the primary alignment.We filter the probes by 50 % samples with FPKM > 0.2 and filter the samples by hclust in R (similar with the HG1.0 /HG1.1 part). In case of multi-probe-per-gene, we take the max of the probe-based values as the representative values of the gene.
Differential expression analysis
After the basic pre-processing, data were performed the unified adjustment and differential expression analyses pipelines. The linear regression-based adjustment for the chosen covariates and the potential covariates detected by svafunction. Differential expression among different ages was assessed using the lm function in R, with the following inputs: the adjusted gene expression matrix and the age information. We got the differential expression gene list in sex and brain regions by Students’ t-test and Analysis of variance (ANOVA) respectively. The resulting P values were then adjusted form multiple hypothesis testing using false discovery rate (FDR) estimation, and the differential expressed genes were determined as those with an estimated FDR < 0.05. FDR was calculated by qvalue in R (The code can be downloaded from https://github.com/ChuanJ/DEGlist.R). In the table, *** means FDR < 0.001, ** means 0.001 ≤F DR < 0.01, * means 0.01 ≤F DR < 0.05, - means FDR > 0.05. Note: The results are based on all the data in the platform you selected and are not specific to the certain age range or brain region.
Co-expression analysis
We applied weighted gene co-expression network analysis (WGCNA) to the matrix of pairwise gene co-expression values. WGCNA recovers a network that consists of nodes (genes) and edges connecting nodes (i.e., the degree of co-expression for a pair of genes, measured as their correlation after transformation by raising the value to a power, β, that results in an overall scale-free topology). It divides the network into subnetworks called modules, or clusters of genes with more highly correlated expression. The power of 8 platforms is shown below:
Table S2. The power value we chose in the WGCNA analysis
| # | Platform | Power |
|---|---|---|
| 1 | HG-U133P | 3 |
| 2 | HG-U133A | 4 |
| 3 | HuGene-1.0-st | 5 |
| 4 | HuGene-1.1-st | 3 |
| 5 | Illumina Human 49k Oligo Array | 8 |
| 6 | Illumina Human HT-12 V3.0 expression beadchip | 5 |
| 7 | RNA-seq (PloyA+) | 5 |
| 8 | RNA-seq (Ribo-Zero) | 2 |
Note: 49K means Illumina Human 49K Oligo array (HEEBO-7 set), V3.0 means Illumina Human HT-12 V3.0 expression beadchip.
We constructed gene co-expression networks separately in platforms. The connectivity metric between a pair of genes i and j, or kij, is a transformed correlation between their expression profiles, with the matrix A = (kij) known as the unsigned adjacency matrix. kij is defined as |rij|β, using the absolute value of rij, the Pearson correlation coefficient between the profiles of genes i and j, and β is the parameter of a power function. To explore the modular structures of the co-expression network, the adjacency matrix is further transformed into a topological overlap matrix. Use of the topological overlap metric leads to more cohesive and biologically meaningful modules, since it not only represents the direct correlation between two genes but also incorporates their indirect interactions through other genes in the network. Next, to identify discrete modules of highly coregulated genes, average linkage hierarchical clustering of the genes is performed, followed by a dynamic tree-cut algorithm to dynamically cut clustering dendrogram branches into discrete subsets of gene modules. Ordered from largest (the module containing the most genes) to smallest, each module is sequentially assigned.
☆ Data summary
Here is the detailed sample information.
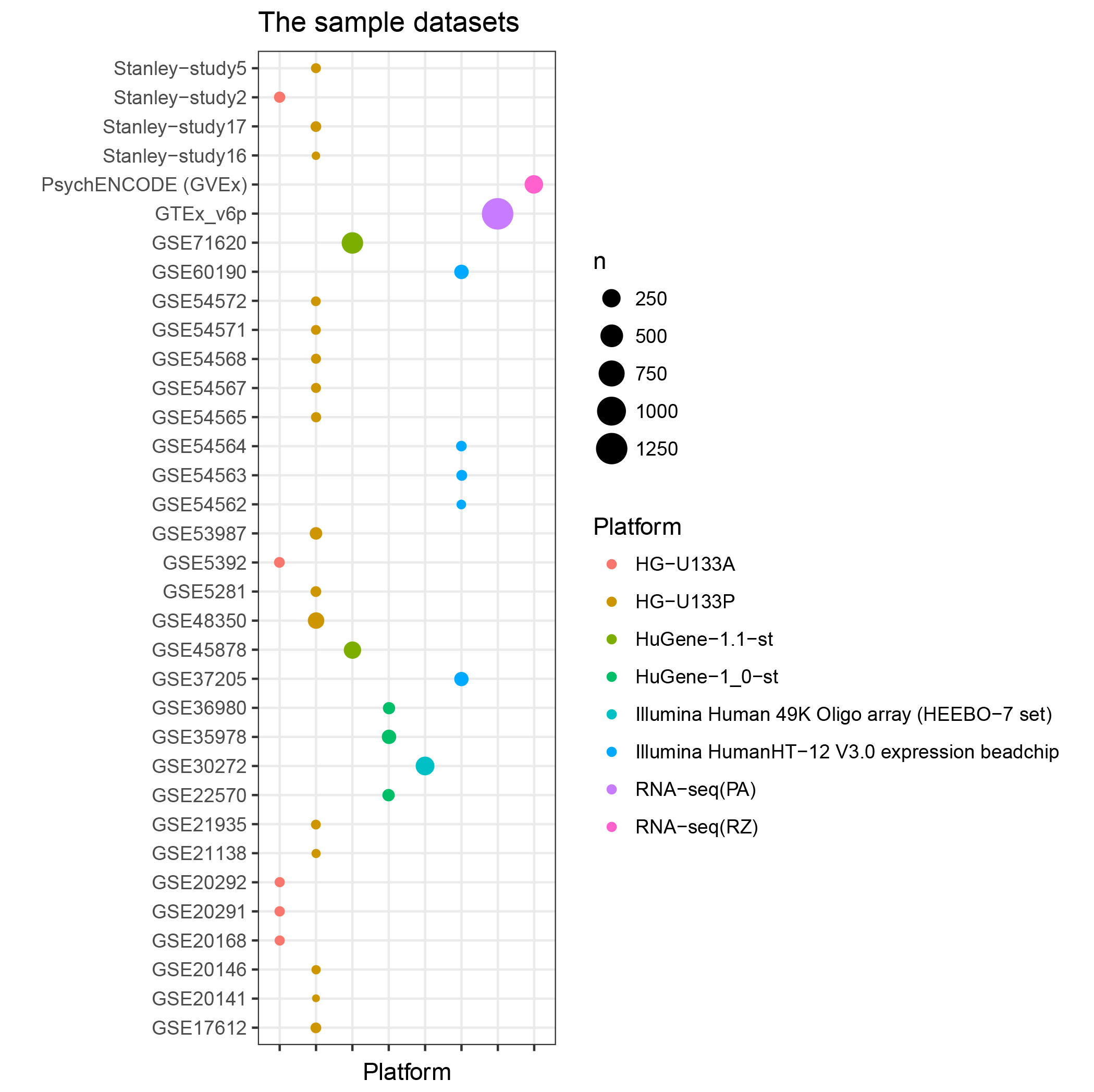
Table S3. The numbers of gene and samples from each platform.
| # | Platform | # of gene | # of samples |
|---|---|---|---|
| 1 | HG-U133P | 21,154 | 402 |
| 2 | HG-U133A | 12,048 | 98 |
| 3 | HuGene-1.0-st | 10,015 | 196 |
| 4 | HuGene-1.1-st | 9,575 | 626 |
| 5 | Illumina Human 49k Oligo Array | 17,141 | 269 |
| 6 | Illumina Human HT-12 V3.0 expression beadchip | 25,123 | 254 |
| 7 | RNA-seq (Ploy A library) | 20,948 | 1258 |
| 8 | RNA-seq (RiboZero) | 21,866 | 258 |
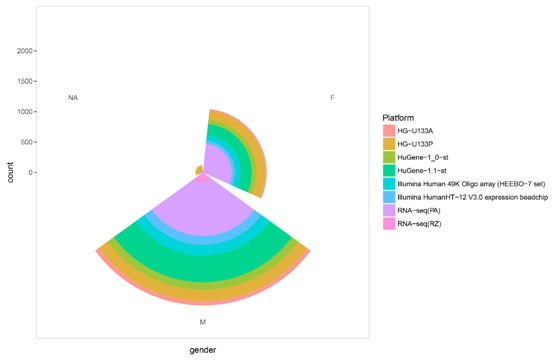
Figure S2. The sample sex distribution in each platform.
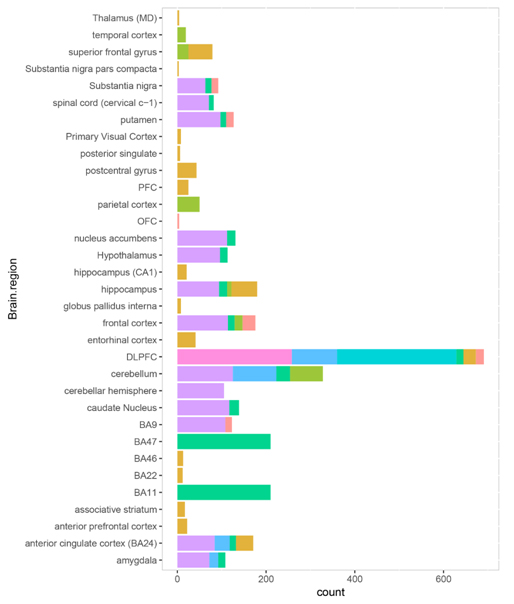
Figure S3. The sample brain region distribution in each platform.
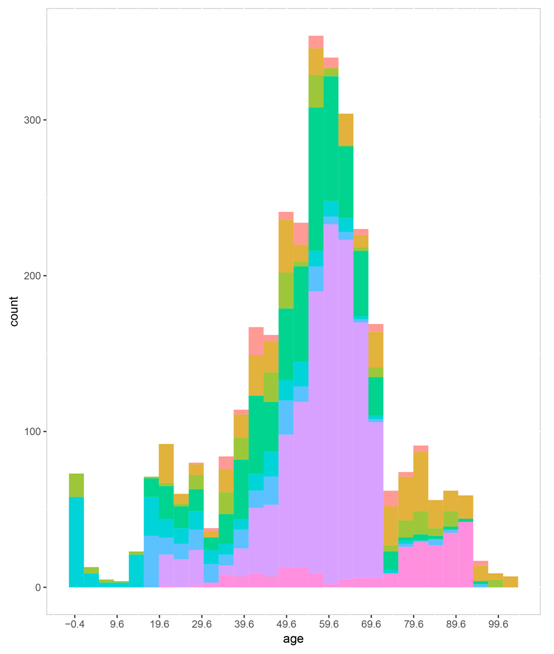
Figure S4. The sample age distribution in each platform.
3. Search for data
☆ Quick search
If you want to scan or browse a single gene’s expression value and its correlation with other genes in brain rapidly, you can use a quick search. You can search the gene by gene symbol, gene ID, or ensemble ID. In the quick search, we have default values on the retrieval restriction. For sex, age, and brain region modules, the default values are “select all”. For platform module, the default value is “RNA-seq (PolyA+).” You can also download the search results.
☆ Search strategy:
Gene symbol: Vague search and automatic matching search are supported. For example, the search is not case-sensitive, if you enter "SCN" or "scn," all of the genes whose gene symbol containing uppercase, lowercase or a mixed case of "SCN" will be listed in the results. When you enter “DRD” or “drd,” all of the genes whose name begin with “DRD” or “drd,” such as “DRD1”, “DRD2”, “DRD4”, “DRD5” will be listed for the remainder.
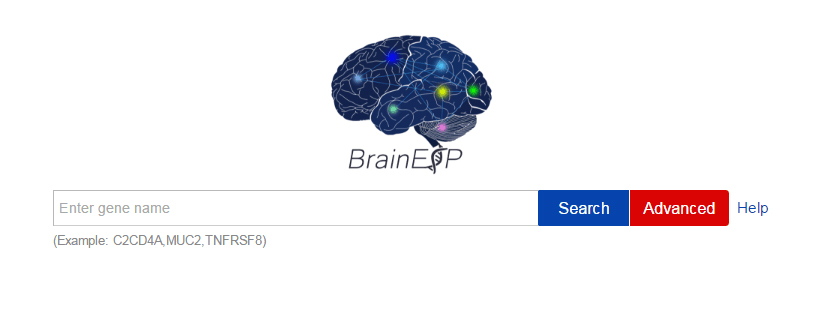
Figure S5. The quick search page with the help and advanced button.
☆ Advanced search
If you are interested in a list of genes, or the gene expression for only a particular age, brain region, or more restrictions, you can select the advanced search. You can search any gene in the human brain for age, sex, brain region, or platform.When you input multiple genes, each item should be separated by acomma.In the age module, you can select any numbers from -0.5 to 110 (ys), such as “20~30” or “-0.5~0”. The negative number indicate the samples are from the embryonic and fetal individuals. In the brain region module, you can select one or more brain regions at a time.In the platform module, if you select "HG-U133P", related data in BrainEXP will be displayed. If you don’t choose a platform, it will have an “RNA-seq (PolyA+)” default. You can also download all the search results.
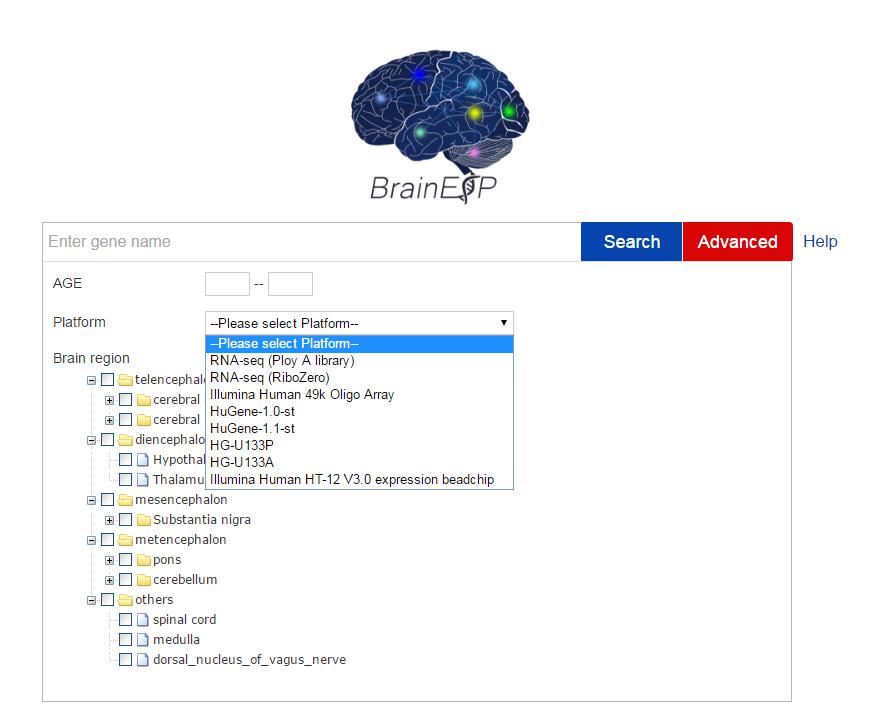
Figure S6. The advanced search page with the age range, platform, and brain region chosen.
4. BrainEXP search results
You can obtainthe gene basic information and gene ontology at first glance when you open the search page. The basic information of gene are get from NCBI.
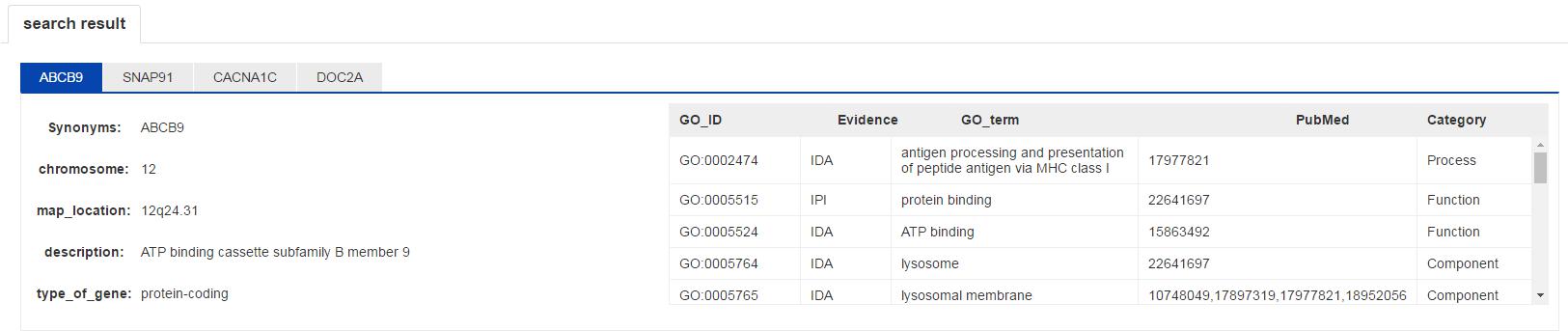
Figure S7. The gene information interface.
This detailed search results contains "Spatiotemporal expression variations" and "Gene co-expression."
☆ Spatiotemporal expression variations
Basic spatiotemporal expression variation information includes differential expression on sex, age, and brain region. See detailsbelow:
Differential expression analysis
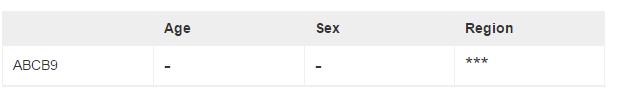
Figure S8. The differential expression analysis results. *** means FDR < 0.001, ** means 0.001 ≤ FDR < 0.01, *means 0.01 ≤ FDR < 0.05, - means FDR > 0.05. Note: The results are based on all the data in the platform you selected and are not specific to the certain age range or brain region.
Gene expression in different brain regions and sexes
This part displays a box plot by calling all the expression data in BrainEXP. It can exhibit brain gene expression level difference among different brain regions and sexes.
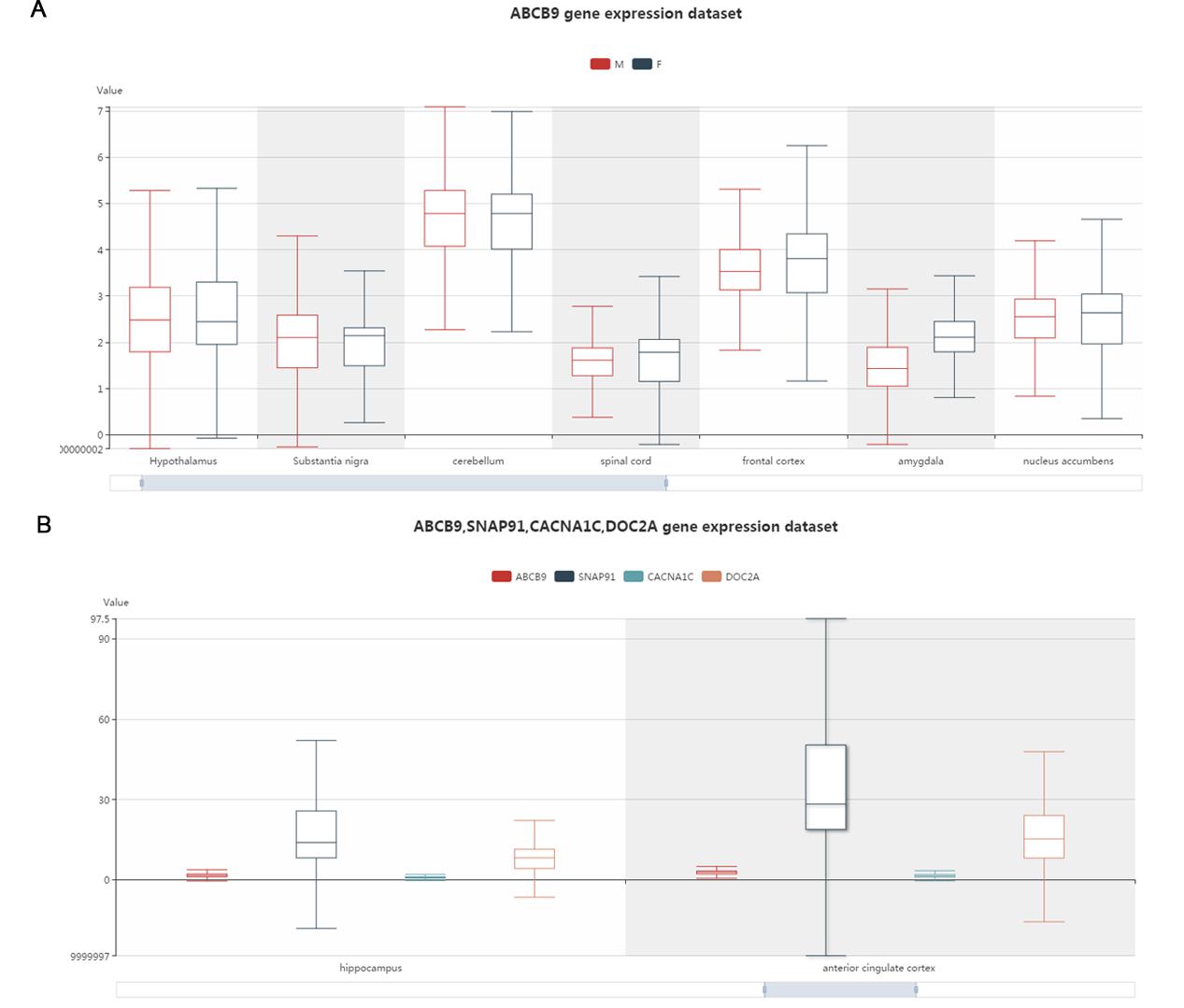
Figure S9. The gene expression in different brain regions and sex interface.You can drag the image until the image is the size that you want. A. the one gene search results. The different colors represent different sexes. B. Multiple genes search results. The different colors represent different genes.
Gene expression in different brain regions and ages
This part shows a scatter plot by calling all the expression data in BrainEXP. It can exhibit brain gene expression level among different agesand brain regions. See the graph below.
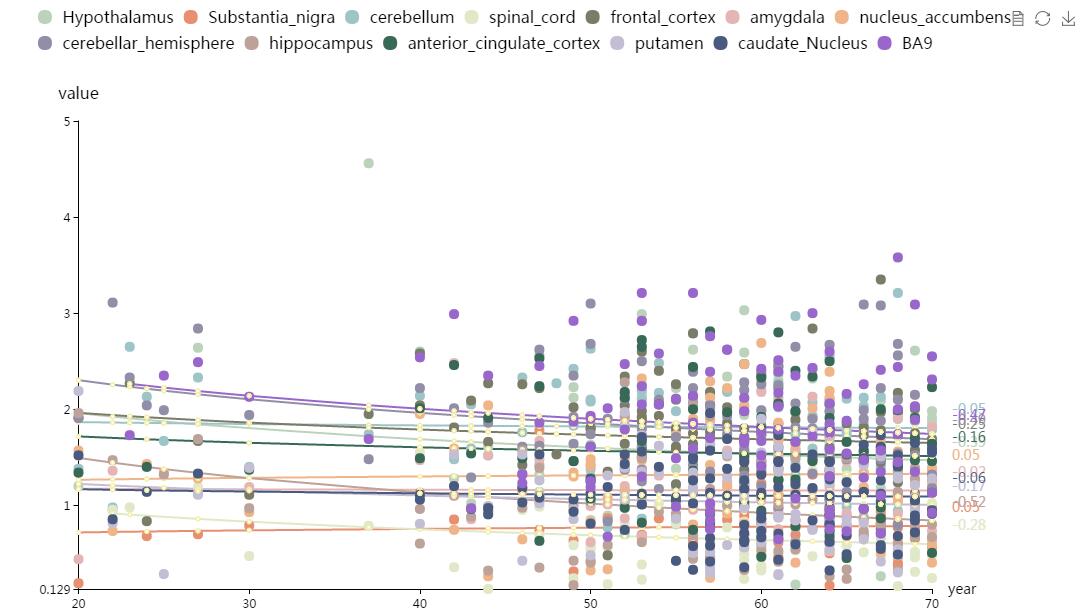
Figure S10. The Gene expression in different brain regions and sex interface. You can click the legend to delete the related points. The number sideward means the gradient of the trend line. The different colors represent different brain regions.
☆ Gene co-expression
We applied WGCNA to createthe matrix of gene co-expression values. The results are displayed through four different modes. The first is the co-expression gene network. Gene nodes connect if the corresponding genes significantly co-expressed across samples at each platform. We choose the top 10 co-expressed genes ordered by the adjacency (the higher the absolute adjacency value, the stronger the connection will be) (Fig.S11). Second, the co-expression pattern shows the other gene expression levels with the similar pattern (Fig.S12). Third, the correlation of co-expression genes gives the correlation coefficients of these 11 genes (top 10 genes plus the query) displayed by the heat map (Fig.S13). Finally, the WGCNA cluster dendrogram will tell you in which module the query gene is detected and how many genes are in the same module (Fig.S14). See graph below.
Co-expression gene network
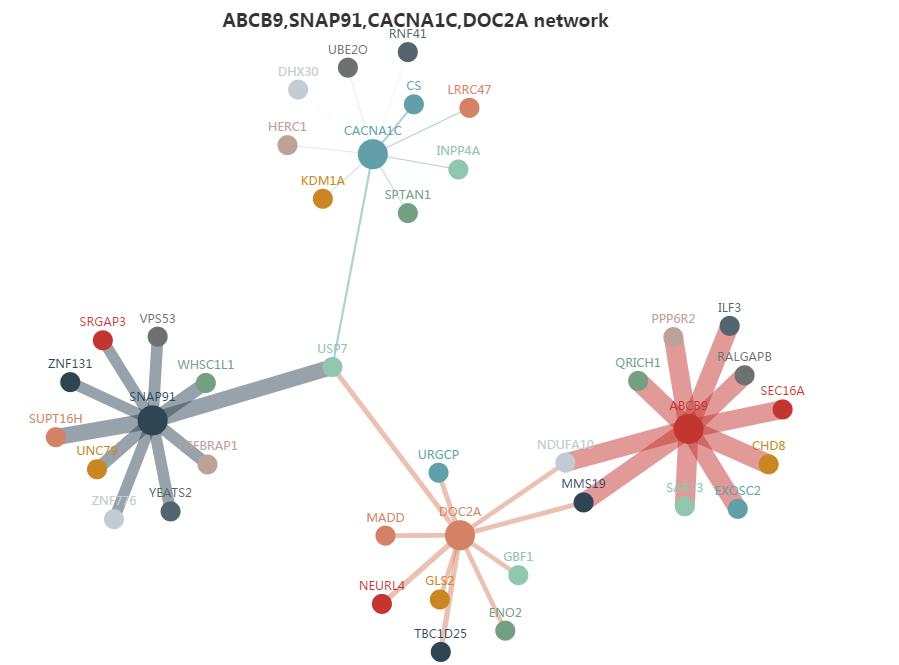
Figure S11. The network constructed by the top 10 gene searched related. The different colors represent different genes. This is ordered by the weight-value calculated by WGCNA. The more related with each other, the line will be much thicker.
Co-expression pattern
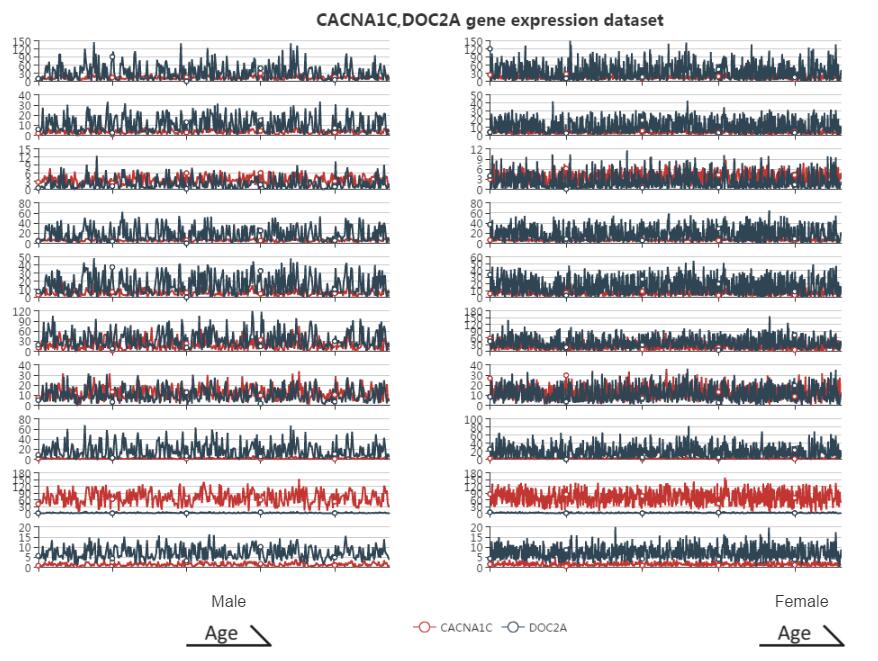
Figure S12. The co-expression pattern interface. Users can get the expression degree of the related search gene. The x-axis represents the samples in the searched platform. The left is the male expression pattern, and the right is female. Expression pattern plot in each part is ordered by age so that the users can see the similarity of co-expression genes pattern. The y-axis represents the expression value of each related gene. The different color represents different related gene.
Correlation of co-expression gene
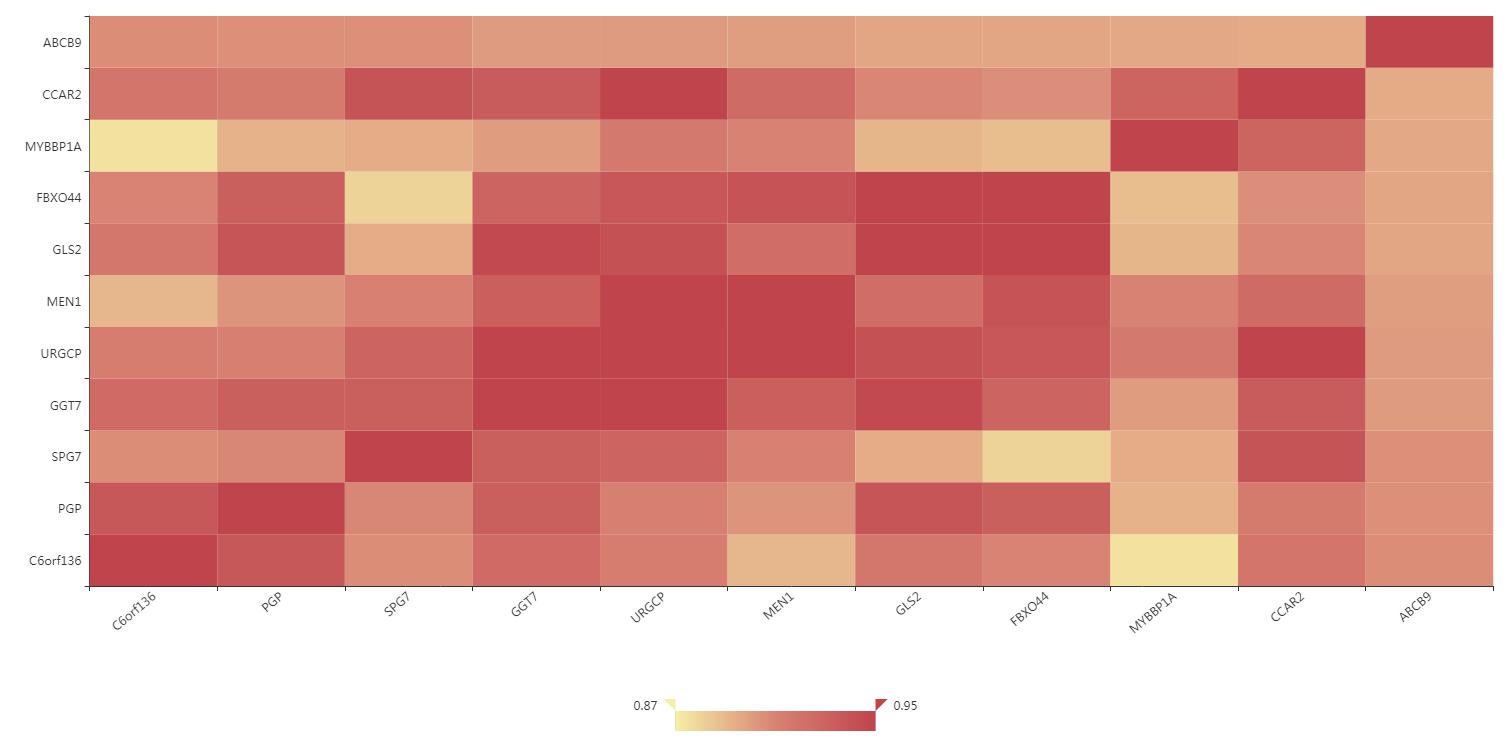
Figure S13. The correlation of co-expression gene interface.The different colors represent different degrees of correlation value. Users can click the cells to get detailed correlation values.
WGCNA cluster dendrogram
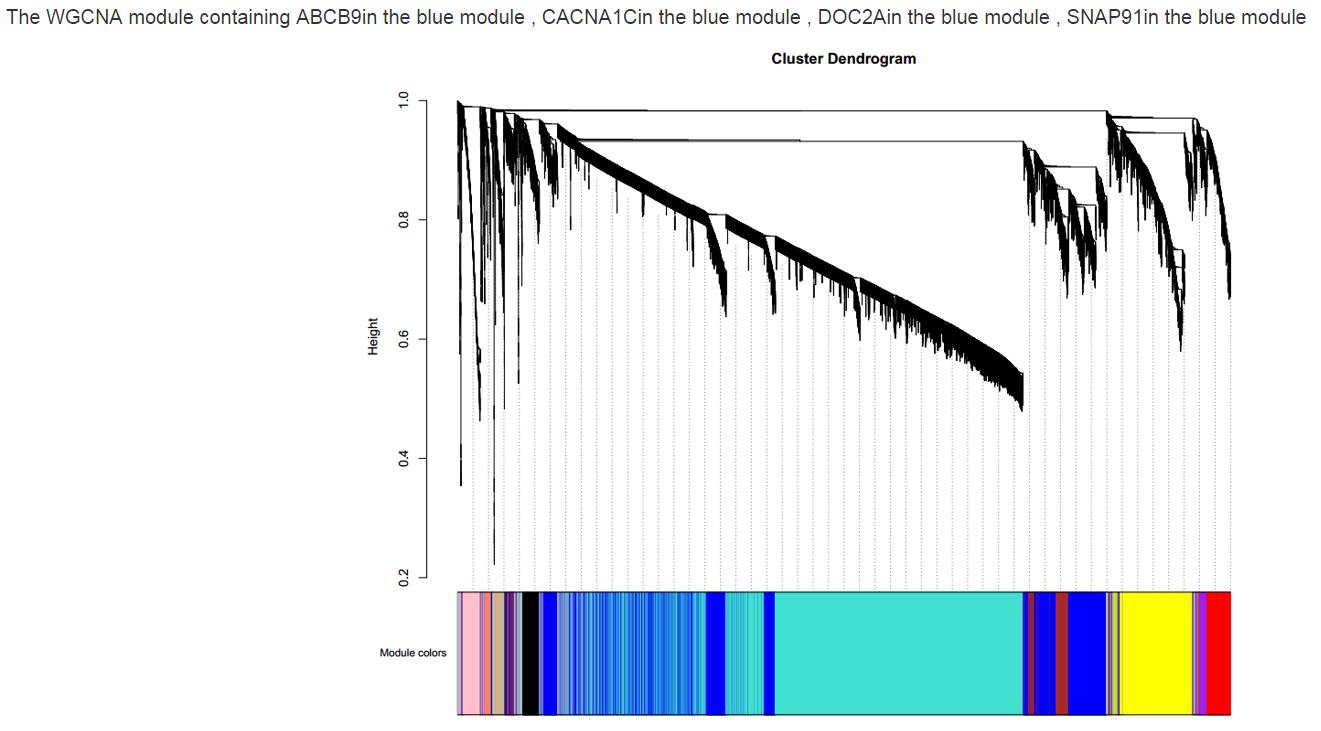
Figure S14. The WGCNA cluster dendrogram interface.The different colors represent different modules. The chart shows in which module the gene was detected. Users can see about how many genes are in the same module with the gene of interest.
5. Note
If you get an empty list or graph after submitting a gene or gene list, it may be due to your restriction of retrieval is too strict, or the gene you submittedhas been filtered during the pre-processing. Again, you are suggested to restrict to a particular platform because different platforms have different sample distributions.
Table S4. The referenced periods from brainspan.
| Period | Description | Age | Age by Y |
|---|---|---|---|
| 1 | Embryonic | 4 PCW ≤ Age < 8 PCW | -0.08 ≤ Age < -0.15 |
| 2 | Early fetal | 8 PCW ≤ Age < 10 PCW | -0.15 ≤ Age < -0.19 |
| 3 | Early fetal | 10 PCW ≤ Age < 13 PCW | -0.19 ≤ Age < -0.25 |
| 4 | Early mid-fetal | 13 PCW ≤ Age < 16 PCW | -0.25 ≤ Age < -0.31 |
| 5 | Early mid-fetal | 16 PCW ≤ Age < 19 PCW | -0.31 ≤ Age < -0.36 |
| 6 | Late mid-fetal | 19 PCW ≤ Age < 24 PCW | -0.36 ≤ Age < -0.46 |
| 7 | Late fetal | 24 PCW ≤ Age < 38 PCW | -0.46 ≤ Age < -0.73 |
| 8 | Neonatal and early infancy | 0 M (birth) ≤ Age < 6 M | 0 ≤ Age < 0.5 |
| 9 | Late infancy | 6 M ≤ Age < 12 M | 0.5 ≤ Age < 1 |
| 10 | Early childhood | 1Y ≤ Age < 6 Y | 1 ≤ Age < 6 |
| 11 | Middle and late childhood | 6Y ≤ Age < 12 Y | 6 ≤ Age < 12 |
| 12 | Adolescence | 12Y ≤ Age < 20 Y | 12 ≤ Age < 20 |
| 13 | Young adulthood | 20Y ≤ Age < 40 Y | 20 ≤ Age < 40 |
| 14 | Middle adulthood | 40Y ≤ Age < 60 Y | 40 ≤ Age < 60 |
| 15 | Late adulthood | 60Y ≤ Age | 60 ≤ Age |
6. Q & A
Q: How do I choose a platform in the advanced search?
A: It depends on your research and our sample information. If you want to search gene expression in multiple brain regions, you are advised to choose RNA-seq (PA). If you want the transcript expression in a specific brain region, such as frontal cortex, you are advised to choose RNA-seq (RZ). If you want fetal gene expression data, Illumina Human 49K Oligo array (HEEBO-7 set) is recommended. If you want to compare the expression levels with your own study, you can choose the corresponding platforms you used.
The supplementary sample information to help you choose a platform.
| Platform | F | M | NA | Total |
|---|---|---|---|---|
| HG-U133A | 28 | 70 | 98 | |
| HG-U133P | 146 | 185 | 71 | 402 |
| HuGene-1.1-st | 184 | 442 | 626 | |
| HuGene-1_0-st | 71 | 125 | 196 | |
| Illumina Human 49K Oligo array (HEEBO-7 set) | 91 | 177 | 1 | 269 |
| Illumina HumanHT-12 V3.0 expression beadchip | 54 | 146 | 54 | 254 |
| RNA-seq(PloyA+) | 385 | 873 | 1258 | |
| RNA-seq(Ribo-Zero) | 89 | 169 | 258 | |
| Total | 1048 | 2187 | 126 | 3361 |
Q: How about the influence of race?
A: Race indeed impacts the gene expression level. The main race in our data is Caucasian European, only 380 samples are black. We did not separate samples based on racein this version.
Q: Are there some samples from the same individual?
A: Yes,but these samples are distributed across the different platforms. It will not impact your search.
Q: Why do you have so manytypes of brain regions?
A: We integrate brain regions from several studies, which have different types of brain regions. Therefore, we have many types of brain regions in BrainEXP.
7.Contact us
If you have any questions, please do not hesitate to contact Chuan Jiao (jiaochuan@sklmg.edu.cn).